AUTOPHAGY REGULATING PATHWAYS
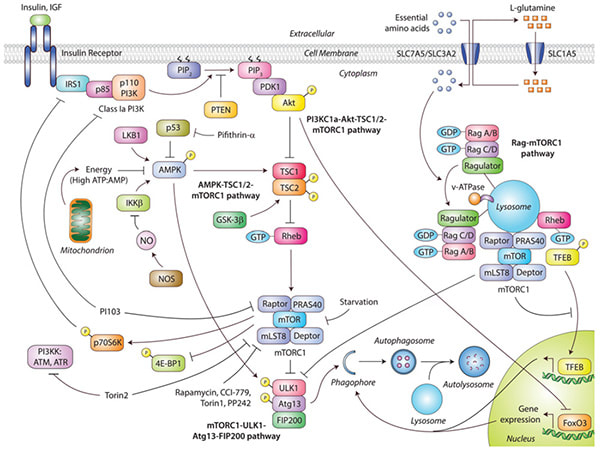
mTOR-dependent autophagy pathways.
(Sarkar S. Biochemical Society Transactions, 2013) Diverse signals like amino acids, growth factors, energy status and stressors activate the mechanistic target of rapamycin complex 1 (mTORC1), which negatively regulates autophagy. Insulin and growth factors act through the PI3KC1a/Akt/TSC/mTORC1 pathway by binding to their cell-surface receptors that activate PI3KC1a and Akt, leading to inhibition of TSC1/2, thereby allowing Rheb to activate mTORC1 and inhibit autophagy. Activation of p70S6K by mTORC1 also exerts a feedback loop by inhibiting IRS1 (insulin receptor substrate 1). In addition, activated Akt inhibits FoxO3-mediated transcription of autophagy genes. Influx of amino acids by their cell-surface transporters function through the Rag/mTORC1 pathway by activating Rag GTPases bound to the lysosome-resident Ragulator, thereby recruiting mTORC1 on the lysosomal surface where Rheb causes its activation, leading to suppression of autophagy. Lysosome-localized activated mTORC1 also sequesters TFEB to prevent its nuclear translocation and transcription of autophagy and lysosomal genes. Energy status and stress signals act through the AMPK/TSC/mTORC1 pathway to modulate autophagy. High ATP/AMP ratio, NO or cytoplasmic p53 inhibits AMPK, thereby preventing TSC1/2 activation that causes Rheb to activate mTORC1 and inhibit autophagy. The ULK1–Atg13–FIP200 complex regulates autophagosome synthesis downstream of mTORC1. Inhibition of mTORC1 during starvation or pharmacologically with rapamycin, CCI-779, Torin1 or PP242 stimulates autophagy. Dual inhibition of mTORC1 and PI3KK with Torin2, or PI3KC1a and mTORC1 with PI103, also activates autophagy. ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia- and Rad3-related; IGF, insulin-like growth factor; PIP2, PtdIns(4,5)P2; PIP3, PtdIns(1,4,5)P3; PRAS40, proline-rich Akt substrate of 40kDa. |
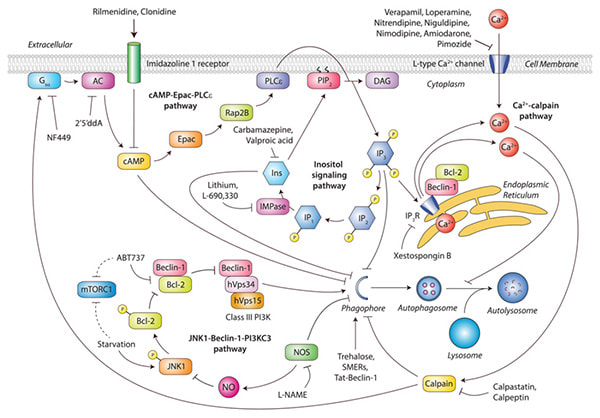
mTOR-independent autophagy pathways.
(Sarkar S. Biochemical Society Transactions, 2013) Various signalling cascades involving cAMP/Epac/PLCε, Ca2+/calpain and inositol (Ins) signalling pathways regulate autophagy independently of mechanistic target of rapamycin complex 1 (mTORC1). Elevation of cAMP levels by adenylate cyclase (AC) activates Epac that in turn activates Rap2B, leading to PLCε-mediated hydrolysis of PtdIns(4,5)P2 (PIP2) to generate Ins(1,4,5)P3 (IP3), which inhibits autophagy. Ins(1,4,5)P3 binds to IP3Rs on the ER to release stored Ca2+ that impairs autophagosome maturation by blocking autophagosome–lysosome fusion. In addition, an increase in cytosolic Ca2+ levels due to Ca2+ efflux from ER stores as well as Ca2+ influx through L-type Ca2+ channels increases its cytosolic levels, resulting inactivation of calpain which inhibits autophagy. Calpain activates Gsα, increasing adenylate cyclase (AC) activity that generates cAMP to suppress autophagy, thereby creating an autophagy-inhibitory link between these pathways. Multiple drug targets acting at distinct stages of these mTOR-independent signalling pathways induce autophagy, such as IMPase inhibitors (lithium, L-690,330), inositol-lowering agents (carbamazepine, valproicacid), Ca2+ channel blockers (verapamil, loperamide, nitrendipine, niguldipine, nimodipine, amiodarone or pimozide), calpain inhibitors (calpastatin, calpeptin), Gsα inhibitor (NF449), adenylate cyclase inhibitor (2’5’ddA), imidazoline receptor agonists (rilmenidine, clonidine) and IP3R antagonist (xestospongin B). Other mTOR-independent autophagy enhancers include trehalose, SMERs and Tat-Beclin-1. The NOS inhibitor L-NAME also stimulates autophagy, whereas NO generated by the NOS isoforms suppresses autophagy by inhibiting the JNK1/Beclin-1/PI3KC3 pathway. Binding of Bcl-2 to Beclin-1 inhibits autophagy, whereas BH3 mimetics like ABT737 induces autophagy by preventing this interaction. Starvation phosphorylates JNK1, which in turn causes Bcl-2 phosphorylation, thereby disrupting Beclin-1–Bcl-2 interaction to promote the Beclin-1–hVps34 autophagy-initiating complex. The PI3KC3 complex comprising Beclin-1–hVps34–hVps15 regulates autophagosome synthesis possibly downstream of the mTOR-independent pathways, although mechanistic details remain to be elucidated. Note that starvation and ABT737 also inhibit mTORC1 activity. DAG, diacylglycerol. |
AUTOPHAGY MODULATORS

AUTOPHAGY SCREENING PLATFORMS
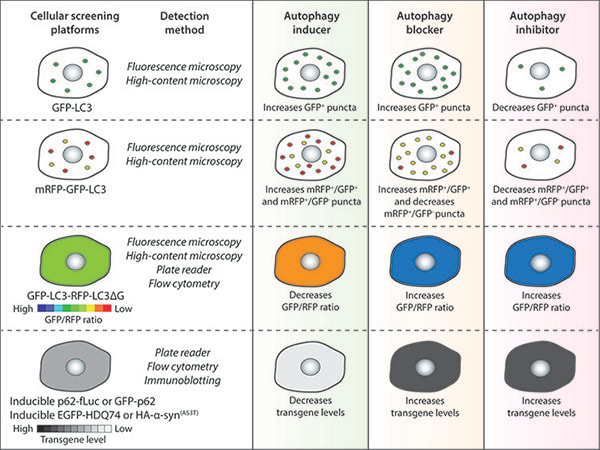
Autophagy chemical screening strategies in mammalian cells.
(Panda P.K. et al. Frontiers in Cell and Developmental Biology, 2019) Chemical screening methods that are commonly used for identifying autophagy modulators are based on autophagy reporters (LC3) or autophagy substrates (p62 or aggregation-prone proteins). The detection methods for the respective assays and the expected readouts for autophagy inducers, blockers or inhibitors are indicated as a general guidance. |
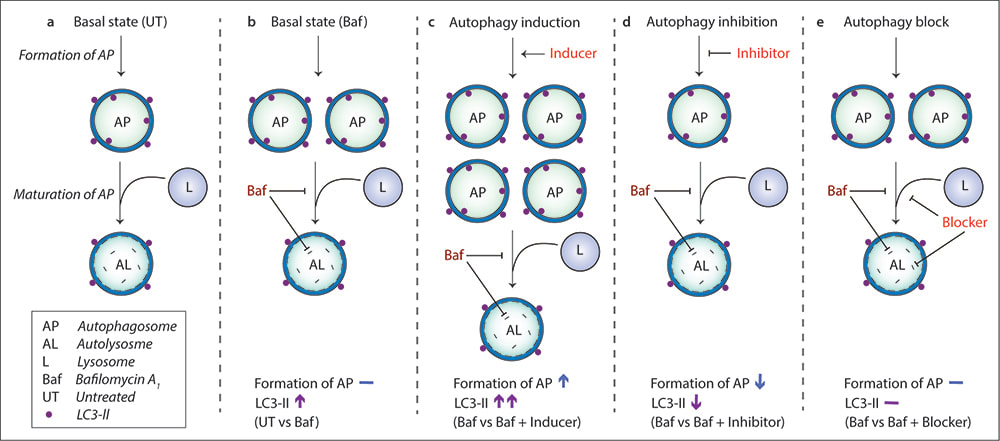
Autophagosome formation assay with bafilomycin A1for characterizing autophagy modulators.
(Seranova E. et al. Methods in Molecular Biology, 2019) In basal state (untreated condition), autophagosome formation and maturation occur under basal autophagy (a). To determine the effects of autophagy modulators on autophagosome formation, the autophagy pathway is first clamped with a saturating concentration of Bafilomycin A1 (Baf), which accumulates autophagosomes (LC3-II levels) by preventing lysosomal cargo degradation and autophagosome maturation (b). An autophagy inducer (which normally increases autophagosomes on its own) will cause greater accumulation of autophagosomes in Baf-treated cells compared to Baf treatment alone due to increased autophagosome formation (c). An autophagy inhibitor (which normally decreases autophagosomes on its own) will cause lesser accumulation of autophagosomes in Baf-treated cells compared to Baf treatment alone due to decreased autophagosome formation (d). An autophagy blocker (which normally increases autophagosomes on its own) will have no change in the number of autophagosomes in Baf-treated cells compared to Baf treatment alone because Baf causes maximal block in autophagy (e). |
|
|
|
